Endocrinopatias

A hiperprolactinemia é a síndrome caracterizada pelo excesso de produção de prolactina, apresentando pico de prevalência entre 25 e 34 anos no sexo feminino. Ao contrário de outros hormônios, o hipotálamo exerce controle inibitório sobre a secreção desse hormônio nos lactotrofos da adenohipófise, mediado pela dopamina circulante no sistema porta-hipofisário.
Clinicamente, a hiperprolactinemia pode manifestar-se com infertilidade, diminuição da libido, galactorreia e osteoporose, através da ação inibidora sobre a secreção das gonadotrofinas hipofisárias. Nos homens, cursa também com ginecomastia e disfunção erétil e, nas mulheres, alteração do ciclo menstrual e hirsutismo.
As causas fisiológicas costumam cursar com valores hormonais inferiores a 50ng/ml (exceto gravidez), como na lactação, estresse/sono, exercício, estimulação mamária/coito e estimulação torácica (trauma, queimadura).
O uso de fármacos é a principal causa de hiperprolactinemia não fisiológica, com destaque para os anticonvulsivantes, anestésicos, opioides, antidepressivos, estrogênios e alguns anti-hipertensivos, como verapamil e metildopa. Fármacos neurolépticos interferem na ação da dopamina no sistema porta-hipofisário, responsável pela regulação inibitória da secreção de prolactina. Nesses casos, a suspensão do medicamento corrige o distúrbio.
Outras causas incluem doenças sistêmicas como hipotireoidismo primário, insuficiência hepática e renal, epilepsia e doença ovariana policística, a síndrome da sela vazia e a acromegalia.
Níveis hormonais superiores a 150-200ng/ml sugerem prolactinomas, sendo a segunda causa patológica mais frequente. Outros tumores intrasselares podem comprimir a haste hipofisária e resultar em valores elevados de prolactina. Meningiomas, gliomas, tumores metastáticos e craniofaringiomas são alguns exemplos, sendo chamados de “pseudoprolactinomas”.
O diagnóstico laboratorial exige pelo menos duas dosagens de prolactina séricas basais elevadas. A dosagem de outros elementos pode ser necessária conforme a causa aparente do distúrbio, como FSH e LH, estrogênio, testosterona, TSH e T4 livre e avaliação da função renal e hepática. A investigação radiológica auxilia o diagnóstico de massas selares.
Macroprolactinomas de grande extensão podem cursar com níveis baixos de prolactina em ensaio imunométrico pela saturação dos anticorpos de captura e revelação do método (efeito gancho). Neste caso, realiza-se a dosagem de prolactina com diluição prévia do soro.
Em pacientes com diagnóstico de hiperprolactinemia, porém sem sinais clínicos, a pesquisa de macroprolactina em gel de polietilenoglicol ou cromatografia líquida é fundamental para evitar o diagnóstico errôneo de um tumor. A estrutura da macroprolactina é, geralmente, relacionada à presença de imunoglobulinas IgG que se ligam à prolactina e alteram sua capacidade de ligação aos receptores específicos, aumentando sua meia-vida em circulação.
O uso de agonistas dopaminérgicos (Cabergolina, Bromocriptina) tem obtido sucesso na redução de até 80% do tamanho de prolactinomas e em casos de hiperprolactinemia idiopática. A cirurgia é indicada em casos de pseudoprolactinoma, macroprolactinomas com complicações, crescimento tumoral e falha no tratamento medicamentoso após 3 meses.
# HIPERPROLACTINEMIA
# HIPOPITUITARISMO
Hipopituitarismo é a diminuição da função hipofisária, que pode ser hormonal seletiva ou comprometer a secreção de todos os hormônios (pan-hipopituitarismo). A síndrome clínica varia de acordo com o tipo celular mais envolvido.
A falência hormonal da hipófise anterior costuma obedecer a uma sequência: GH > LH/FSH > TSH > ACTH > PRL. Pode ser decorrente de um problema hipofisário ou hipotalâmico, sendo que lesões hipotalâmicas ou pediculares tendem a elevar os níveis de prolactina por comprometer a influência inibitória sobre os lactotrofos. A deficiência de prolactina só ocorre quando a destruição hipofisária é muito grave.
A causa mais comum de pan-hipopituitarismo é o adenoma hipofisário, com disfunção progressiva da secreção hormonal. Diversas outras patologias podem comprometer a hipófise como a Síndrome da Sela Vazia, Síndrome de Sheehan (necrose hipofisária periparto), anemia falciforme, amiloidose, hemocromatose, tuberculose, infecções fúngicas e radioterapia.
Os núcleos médio-basais do hipotálamo contêm os neurônios responsáveis pela secreção dos fatores hipofisiotrópicos e se localizam próximo aos núcleos supraóptico e paraventricular que produzem ADH e ocitocina. Uma única lesão pode comprometer toda a região, levando ao pan-hipopituitarismo, diabetes insipidus central e, eventualmente, obesidade hipotalâmica. Os tumores que comprometem o hipotálamo estão fortemente associados, sendo mais comum o adenoma hipofisário com invasão suprasselar.
O craniofaringioma é o segundo mais comum, mais frequente em crianças e com tendência à calcificação, identificada na TC ou RNM. Manifesta-se com sinais de lesão expansiva do SNC (cefaleia, vômitos, alterações visuais), poliúria e pan-hipopituitarismo. Outras causas hipotalâmicas são: embriopatias da linha mediana, radioterapia, sarcoidese, Síndrome de Prader-Willi e Síndrome de Kallman.
No adulto, o quadro clínico inicial mais frequente é o hipogonadismo, enquanto na criança o déficit de crescimento é a apresentação habitual. Em geral, os sintomas podem incluir sensação de cansaço, perda de peso, infertilidade, intolerância ao frio, diminuição do apetite e queda de pelos.
Diante da suspeita de hipopituitarismo, é importante determinar o grau de deficiência hormonal e a etiologia. O rastreio consiste nas dosagens basais dos hormônios hipofisários e das glândulas-alvo. Provas funcionais (testes dinâmicos) podem ser usados para complementar a avaliação inicial. A avaliação da região hipotálamo-hipofisária é feita, preferencialmente, pela RNM, sendo que a TC é mais utilizada para definir calcificações.
O tratamento objetiva tratar a causa básica e corrigir os distúrbios hormonais. As reposições são feitas conforme os hormônios deficientes, por meio da Prednisona, L-Tiroxina, estrogênios conjugados (mulheres) e ésteres de testosterona (homens). A reposição de GH em adultos é controversa. Geralmente, o uso medicamentoso é contínuo e exames de sangue são realizados regularmente para verificar os níveis hormonais.
# ADENOMAS HIPOFISÁRIOS
Adenomas hipofisários são neoplasias bem diferenciadas originárias da adenohipófise. Clinicamente, apresentam-se com a síndrome caracterizada pelo excesso do hormônio secretado. Cerca de 25% dos adenomas são não funcionantes e podem ser uma causa de hipopituitarismo.
O tipo mais comum de adenoma hipofisário é o prolactinoma. Geralmente, consiste em um microadenoma (< 10mm), mais comum no sexo feminino, e manifesta-se com hipogonadismo e galactorreia. Níveis de prolactina se correlacionam positivamente com o tamanho do tumor.
Os macroadenomas (> 10mm) são, em sua maioria, do tipo misto, com produção aumentada dos diferentes hormônios da adenohipófise. A clínica varia entre gigantismo/acromegalia (GH), doença de Cushing (ACTH) e puberdade precoce ou pan-hipopituitarismo (LH/FSH). O hipertireoidismo resultado da hipersecreção de TSH é muito raro.
# ADENOMAS HIPOFISÁRIOS
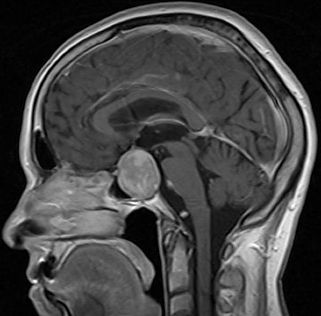
Em geral, o tratamento é conservador e o acompanhamento por exames de imagem é fundamental para avaliar o tamanho do adenoma e se está comprometendo estruturas vizinhas. Neste caso, a ressecção cirúrgica é indicada.
A cirurgia transesfenoidal é o tratamento de escolha para pacientes que não apresentam redução dos valores hormonais com tratamento clínico correto, para os casos de acromegalia e para a doença de Cushing. A chance de cura depende do tamanho do tumor.
Macroadenomas com expansão extrasselar apresentam taxas de cura pela cirurgia muito baixas.
Na preferência pelo tratamento farmacológico, os Análogos da Somatostatina são as drogas de primeira linha para bloquear a hipersecreção de GH, e os Agonistas Dopaminérgicos podem ser utilizados para os excessos de PRL e ACTH. Outros fármacos como o cetoconazol e a metirapona também podem ser indicados em adenomas adrenocorticotróficos no intuito de inibir a esteroidogênese adrenal.
A radioterapia é uma opção para casos refratários ao tratamento medicamentoso, que não apresentam indicação cirúrgica e para recidivas após cirurgia. A radioterapia estereotáxica é uma modalidade mais precisa e apresenta riscos de complicação, como hipopituitarismo e disfunção cognitiva, menores que a técnica convencional. Pode também ser bem indicada em crianças, com taxa de cura de até 80%.
Referência:
MEDCURSO 2010 – volume 2 Endocrinologia – pag 21, 55-62
VIEIRA, J. G. H., Macroprolactinemia. Arq Bras Endocrinol Metab, vol.46 n.1 São Paulo Feb. 2002.
# HIPOTIREOIDISMO

O hipotireoidismo caracteriza-se pela hipofunção da glândula Tireóide, queda dos seus hormônios séricos, causando diversos sintomas de queda do metabolismo. A deficiência de iodo continua sendo a sua causa mais comum em todo o mundo, nas áreas com suficiência de iodo a doença autoimune Tireoidite de Hashimoto e as causas iatrogênicas, como o tratamento do hipertireoidismo, são as causas mais comuns. Chamamos de Hipotireoidismo primário quando a causa acomete a glândula, por exemplo a Tireoidite de Hashimoto, deficiência de iodo, iatrogenia e outros, hipotireoidismo secundário é quando a causa acomete a glândula indiretamente, afetando a secreção de TSH, como por exemplo o hipopituitarismo, traumatismos, tumores hipotalâmicos e outros.
O Hipotireoidismo autoimune (Tireoidite de Hashimoto) te taxa de incidência de 4/1000 mulheres e 1/1000 homens e a prevalência da doença aumenta com a idade. Ocorre pela presença de autoanticorpos (anti-TG e anti-TPO) contra a glândula, gradualmente reduzindo a função tireoidiana, em dado momento existe uma fase de compensação em que o aumento do TSH mantém os níveis normais dos hormônios tireoidianos, este momento é chamado de hipotireoidismo subclínico. Existe uma infiltração linfocítica da glândula com a formação de centros germinativos, atrofia dos folículos tireoidianos, metaplasia, ausência de coloide e fibrose. Os anticorpos anti-TGO e anti-TG são marcadores importantes. As manifestação clínicas são cansaço, fraqueza, pele seca, sensibilidade ao frio, queda de cabelo, constipação, rouquidão, dispneia, parestesia, audição prejudicada, dificuldade de concentração, memória prejudicada, pele seca, mixedema, edema periférico, bradicardia, depressão e outros. O tratamento normalmente consiste em repor os hormônios tireoidianos com Levotiroxina. Uma complicação que pode ocorrer é o coma mixedematoso, que é uma forma de hipotireoidismo descompensado, onde todos os órgãos são afetados, ocorrendo acúmulo de líquido, alteração da contratilidade miocárdica, dificuldade de controle ventilatório, hipercapnia, coma, alterações cognitivas, diminuição da taxa de filtração glomerular, queda da temperatura corporal e outros. O tratamento é repor os hormônios, podendo fazer uma dose de levotiroxina IV de ataque (pode-se utilizar liotironina também), hidrocortisona parenteral e terapia de suporte para manter a temperatura corporal, apoio ventilatório, solução salina hipertônica ou a glicose IV se necessário.
O hipotireoidismo congênito acomete 1/4000 recém-nascidos, podendo estar associado com fatores maternos como a presença de anticorpos antitireoidianos ou anti-TSH no leite, sendo assim transitório. O quadro pode permanecer, caracterizando um hipotireoidismo neonatal que ocorre devido a disgenesia da glândula. O recém-nascido pode apresentar icterícia prolongada, problemas alimentares , hipotonia, língua aumentada de volume, maturação óssea retardada e hérnia umbilical. Menos de 10% dos lactentes são diagnosticados com base na clínica. O teste do pezinho é um dos exames de triagem para o diagnóstico do hipotireoidismo congênito e o tratamento baseia-se em suplementar o T4 em dose adequada, pois os hormônios tireoidianos são necessários em dosagens elevadas nessa fase de vida, principalmente no primeiro, para o desenvolvimento neurológico.
Referência:
JAMESON J.L; WEETMAN A.P. Distúrbios da Glândula Tireóide. In: LONGO D.L; et al. Princípios de Medicina Interna de Harrison. 18ed. New York: McGraw-Hill, 2012, v.2, p.2911-2939.
# HIPERTIREOIDISMO
O Hipertireoidismo caracteriza-se por uma função da Tireóide excessiva que pode levar a um quadro de tireotoxicose, que é o estado de excesso de hormônios tireoidianos. As causas primárias, relacionadas diretamente com a glândula, são a Doença de Graves, Bócio Multinodular tóxico, Adenoma tóxico e outros, as causas secundárias são Adenoma hipofisário secretor de TSH, Resistência ao Hormônio Tireoidiano, tireotoxicose gestacional e outros.
Um quadro de tireotoxicose pode acontecer sem hipertireoidismo, como ocorre em lesão da glândula como em traumatismos, tireoidites, e em caso de uso de amiodarona ou ingestão excessiva de hormônios tireoidianos. A principal causa é a Doença de Graves (60% a 80%).
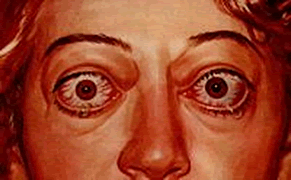
A Doença de Graves está relacionada com fatores genéticos e ambientais, como estresse e a ingestão elevada de iodo. É uma doença auto-imune associada com um autoanticorpo imunoglobulina estimulante da tireóide (TSI) que mimetiza o hormônio TSH em seus receptores, causando um aumento da função da glândula. As manifestações clínicas de tireotoxicose são: fadiga, perda de peso, apetite aumentado, pele quente e úmida, hiperatividade, irritabilidade, disforia, intolerância ao calor, palpitações, diarreia, poliúria, oligomenorréia, perda de libido, insônia, mixedema pré-tibial, perda de massa muscular (devido ao catabolismo aumentado) e outros. A doença de Graves possui uma manifestação específica que é a oftalmopatia de Graves, ocorre por liberação de citocinas pelas células T nos músculos extra-oculares, causando exoftalmia. Na doença de Graves a Tireóide costuma ficar difusamente aumetada para duas ou três vezes seu tamanho normal, com consistência firme, sendo possível algumas vezes auscultar um sopro devido ao aumento da vascularização. O diagnóstico é simples, pela clínica, exame físico da tireoide, dosagem bioquímica dos hormônios para confirmar a tireotoxicose, se necessário pode-se fazer exame laboratorial para identificar os autoanticorpos TSI. O tratamento se dá com agentes antitireoidianos (as tionamidas: propiltiouracil, carbimazol e metilmazol) que inibem a função da tireoperoxidase, tratamento com iodo radioativo e a tireoidectomia.
Referência:
JAMESON J.L; WEETMAN A.P. Distúrbios da Glândula Tireóide. In: LONGO D.L; et al. Princípios de
Medicina Interna de Harrison. 18ed. New York: McGraw-Hill, 2012, v.2, p.2922-2927.
# SÍNDROME DE CUSHING
palmente nas regiões do abdômen, coxas, seios e braços; emagrecimento, pele frágil; cicatrização lenta e acne. Os sintomas também podem variar de acordo com o sexo. Os sintomas mais comuns no sexo feminino são: depósito de gordura no corpo; cabelo facial (hirsutismo) e períodos menstruais irregulares ou ausentes. Enquanto no sexo masculino, os sintomas mais frequentes são: diminuição da libido, diminuição da fertilidade e disfunção erétil. Além disso, outros sintomas podem estar presentes como: fadiga, fraqueza muscular, depressão, ansiedade, irritabilidade, perda de controle emocional, dificuldades cognitivas, hipertensão e perda óssea, levando a fraturas ao longo do tempo.
O diagnostico se faz através de exames complementares, que envolvem testes e dosagens hormonais como, por exemplo: Exame de urina de 24 horas para verificar os níveis de cortisol; Medição dos níveis de cortisol no plasma ou na saliva; Exame de supressão de dexametasona; Dosagem de ACTH no sangue.
O tratamento visa diminuir e estabilizar a concentração de cortisol no organismo. Se a causa da doença for o uso de medicamentos à base de corticosteroide, o médico suspenderá gradualmente seu uso até encontrar outra forma de tratar a doença que necessitava do medicamento. Se a doença for causada por um tumor na glândula suprarrenal, a cirurgia de retirada pode resolver definitivamente o problema. A radioterapia pode ser necessária mesmo depois da cirurgia, mas somente em alguns casos específicos. Caso o tumor não possa ser retirado, o tratamento visará a diminuição dos sintomas e o controle dos níveis de cortisol.

A Síndrome de Cushing é uma patologia desencadeada pela elevada concentração no corpo de hormônio cortisol. Segundo a Organização Mundial da Saúde (OMS), aproximadamente 50 mil pessoas vivem com essa doença atualmente.
A quantidade de cortisol no organismo quando está muito elevada é consequência de dois principais motivos: o uso excessivo de medicamentos corticoides, para tratar doenças reumatológicas, por exemplo. Ou pela excessiva excessiva produção desse hormônio pelo organismo, podendo ser consequência de um tumor nas glândulas, entre outras causas. A apresentação clínica da síndrome de Cushing costuma ser individual e variável. As alterações mais comuns são obesidade e alterações cutâneas progressivas, como por exemplo: depósitos de gordura no corpo; estrias na pele, princi-
# DOENÇA DE ADDISON
A Doença de Addison é o nome dado à condição em que as glândulas suprarrenais não são capazes de sintetizar níveis suficientes de seus hormônios. As glândulas adrenais são divididas em duas partes: o córtex e a medula, com funções diferentes. A primeira produz corticosteroides, como o cortisol, e a segunda produz catecolaminas, como a adrenalina, hormônios esses produzidos em resposta ao estresse. Além de também secretarem a aldosterona, um hormônio diretamente envolvido na regulação da osmalaridade do sangue, e estimulam a conversão de proteínas e gorduras em glicose, ao mesmo tempo em que diminuem a captação da glicose pelas células, aumentando, assim, a utilização de gorduras pelo corpo. O córtex também produz pequenas quantidades de andrógeno, o hormônio sexual masculino, tanto em homens quanto em mulheres. A Doença de Addison pode ocorrer devido a dois grandes motivos distintos, podendo ser classificada de insuficiência adrenal primária e insuficiência adrenal secundária.
Insuficiência adrenal primária: Acontece quando o córtex das glândulas suprarrenais sofre algum tipo de dano, impossibilitando-o de produzir hormônios em quantidades adequadas. Ocorrendo principalmente, devido a um processo autoimune, ou seja, quando as células de defesa do organismo enxergam o córtex adrenal como um agente invasor e atacam-no, prejudicando suas funções. Além de outros motivos, como: Tuberculose, infecções das glândulas suprarrenais, câncer, sangramento das glândulas e uso de medicamentos anticoagulantes.
Insuficiência adrenal secundária: Também pode ocorrer quando o problema inicial é na hipófise. A hipófise é responsável pela produção do hormônio adrenocorticotrófico (ACTH), que estimula o córtex adrenal a produzir seus hormônios. A produção inadequada ou insuficiente de ACTH pode levar a uma queda na produção de hormônios que são normalmente produzidos pelas glândulas suprarrenais, apesar de estas não estarem sendo danificadas por nenhum motivo aparente. Outra causa mais comum para este tipo de insuficiência adrenal é a interrupção abrupta do uso de medicamentos corticoides – bastante comum em pessoas que estão tratando algumas doenças crônicas, como esclerose múltipla e asma.
Geralmente as alterações desenvolvem-se de forma insidiosa, ao longo de vários meses, e podem incluir: astenia, fadiga, perda de peso, diminuição do apetite, hiperpigmentação, hipotensão, hipoglicemia, náuseas, vômitos, diarreia, artralgia, mialgia, irritabilidade, depressão, disfunção sexual em mulheres. Algumas vezes, as alterações podem surgir agudamente, o que configura um caso de insuficiência suprarrenal aguda. Nesses casos, os sinais e sintomas podem incluir: lombalgia, dor abdominal ou nas pernas, vômitos e diarreia severa e consequente desidratação, hipotensão, perda de consciência e hipercalemia.
O diagnostico se faz através de exames complementares, que envolvem testes e dosagens hormonais como, por exemplo: Hemograma; Teste de estimulação do ACTH, que envolve a medição do nível de cortisol no sangue antes e depois de uma injeção de ACTH sintético; Teste hipoglicêmico induzido por insulina, geralmente recomendado para um possível diagnóstico de insuficiência adrenal secundária; Exames de imagem, como tomografia computadorizada e ressonância magnética.
O tratamento se faz através de uma terapia de reposição hormonal. O tratamento consiste na administração de corticosteróides, em alguns casos é necessário, ainda, a associação de um mineralocorticoide, como a fludrocortisona e consiste em um tratamento crônico. Os doentes com doença de Addison devem estar esclarecidos quanto ao risco de insuficiência supra-renal aguda nos casos de infecções, cirurgias ou traumatismos físicos ou psicológicos. Nestes casos devem aumentar a dose de corticosteróides nas doses prescritas pelos médicos assistentes. Devem, ainda, ser portadores de um cartão informativo da sua doença, da terapêutica que fazem, do contacto com o seu médico assistente e dos cuidados a ter em caso de risco. Devido às características particulares desta doença, aos diversos tipos de apresentação clínica e de tratamentos e ao elevado risco de vida que correm estes doentes, recomenda-se a necessidade de avaliações periódicas. A doença de Addison deve ser sempre tratada por endocrinologistas.
# ACROMEGALIA
O termo acromegalia é um neologismo formado por duas palavras gregas: Akron (extremidade) + megalo (grande), referindo-se a uma doença crônica, grave, rara e insidiosa que afeta adultos (na faixa entre 30 e 50 anos), de ambos os sexos, quando o hormônio do crescimento (GH) é produzido em excesso e, consequentemente, produz outro hormônio. Quando o hormônio do crescimento é liberado no sangue, estimula o fígado a produzir o IGF-1 (Insulin Growth Factor), semelhante à insulina tipo 1 que faz crescer músculos, ossos e cartilagens em todo corpo.

Este processo é essencial para o crescimento e reparação dos tecidos do corpo humano. Entretanto, conforme já foi dito, quando isso ocorre na fase adulta, as cartilagens de crescimento já estão fechadas e acabam por deformar os ossos ao invés de alongá-los. Muitas das manifestações clínicas da doença são comuns a outras patologias mais frequentes, o que faz com que o diagnóstico da acromegalia seja muitas vezes tardio, podendo a doença estar ativa sem ser reconhecida durante vários anos (10‐15 anos).
A acromegalia tem uma incidência, aproximada, de 5 casos/milhão/ano e uma prevalência de 60 casos por milhão de pessoas. Os adenomas da hipófise correspondem a 15% de todos os tumores intracranianos, sendo que 90% dos doentes com acromegalia terão um adenoma hipofisário produtor de GH, um macroadenoma em 70% dos casos. Estes tumores podem produzir apenas GH isoladamente ou em associação a outras hormônios, sendo a mais frequente a prolactina (PRL).
As manifestações clínicas da acromegalia são muito variadas, dependem dos níveis de GH e fator de crescimento semelhante à insulina tipo 1 (IGF‐1), da idade do doente, do tamanho do tumor e do tempo decorrido entre o início da doença e o seu diagnóstico. Incluem sinais e sintomas associados ao efeito de massa do tumor hipofisário e efeitos sistêmicos do excesso de GH e IGF‐1, nomeadamente doença cardiovascular, alterações metabólicas, manifestações respiratórias, ósseas e articulares, manifestações gastrointestinais, bem como outras manifestações endócrinas (bócio, hipercalciúria, diminuição da libido, irregularidades menstruais).
As alterações somáticas e da aparência física são muito sugestivas da doença, contudo, podem ter um desenvolvimento muito indolente e insidioso, por vezes ao longo de anos, não sendo, nestes casos, imediatamente perceptíveis. Em alguns doentes, estas alterações fenotípicas são tão sutis que não são percebidas pelo próprio nem pelos seus familiares, sendo também de difícil valorização para o clínico. Mesmo na ausência de características físicas sugestivas da doença, a acromegalia deve ser considerada um diagnóstico diferencial quando coexistem diversas patologias que podem ser explicadas por esta doença.
A acromegalia está associada a um aumento da mortalidade, de cerca de 2‐3 vezes, em relação à população geral, sendo esta, sobretudo, secundária a doença cardiovascular (60%), respiratória (25%) ou neoplásica (15%). Além disso, essa doença aumenta a prevalência de apneia do sono principalmente por estar associada com deformidades craniofaciais, macroglossia e infiltração dos tecidos da via aérea superior. SAHOS (síndrome da apneia e hipoapneia obstrutiva do sono) contribui para o aumento da mortalidade cardiovascular em acromegálicos.
O diagnostico de acromegalia requer a demonstração de que os níveis de GH estão aumentados e desregulados, assim como elevação dos níveis de IGF-1, que refletem a exposição de tecidos periféricos a níveis tonicamente elevados de GH. Nesta doença, a secreção basal de GH está tonicamente elevada com pulsos de liberação mais atenuados. Para fins diagnósticos, um valor menor que 0,04 μg/L referente a uma determinação aleatória dos níveis de GH exclui efetivamente o diagnóstico de acromegalia. Importante ressaltar que a obtenção de valores elevados de GH aferidos de forma aleatória não reflete uma concentração integrada alta desse hormônio. A secreção de GH fica diminuída em cerca de 50% após os 60 anos e também é alterada em função do índice de massa corporal.
Após o diagnóstico clínico-laboratorial de acromegalia, como 99% dos casos são causados por um adenoma hipofisário secretor de GH, está indicada a realização de ressonância magnética (RM) de sela túrcica para identificação e caracterização (dimensões e expansões) do tumor. São feitos cortes sagitais e coronais antes e após a administração IV do gadolíneo. Vários centros do Brasil não dispõem de RM, sendo utilizada a tomografia computadorizada (TC).
Nos pacientes submetidos ao tratamento cirúrgico, é importante que seja realizado o estudo histopatológico para comprovar adenoma, ou seja, excluir a rara possibilidade de hiperplasia somatotrófica, o que nos obrigaria a investigar produção eutópica ou ectópica de GHRH. E também, faz-se a análise imunohistoquímica, através da qual saberemos se o adenoma é produtor apenas de GH ou se também produz PRL. Esta informação será muito importante no momento de decisão terapêutica, pois os adenomas co-secretores de GH e PRL respondem melhor aos agonistas dopaminérgicos do que os adenomas secretores exclusivamente de GH.
A cirurgia transesfenoidal (TSS) é o tratamento primário de escolha para acromegalia, sendo utilizada a via nasal nos pacientes do ambulatório de pesquisa em acromegalia. A terapia medicamentosa atualmente representa a segunda opção de tratamento, após ressecção cirúrgica do adenoma hipofisário. A maioria dos pacientes com acromegalia tem tumores que não são totalmente ressecados, necessitando terapia complementar após a cirurgia para normalizar os níveis de IGF-1 circulantes e manter o GH sérico em níveis seguros. Os análogos de somatostatina, os agonistas dopaminérgicos e os antagonistas do receptor de GH são os grupos de drogas disponíveis para esta finalidade. A radioterapia é, na maioria das vezes, a terceira linha de tratamento da acromegalia, quando as terapias cirúrgica e medicamentosa não são suficientes para diminuir os níveis de GH e IGF-1 para valores seguros.
O diagnóstico o mais precoce possível é fundamental. Atualmente, dispomos de várias opções terapêuticas, as quais devem ser empregadas de maneira agressiva visando o controle da doença. Critérios de cura rigorosos baseados em GH após supressão com glicose e IGF-1 devem ser adotados para avaliação da eficácia do tratamento.
De maneira geral, a manutenção de níveis basais de GH inferiores a 2,5 μg/L após o tratamento se correlaciona com aumento da expectativa de vida para taxas próximas às consideradas normais (controle bioquímico). A maioria dessas comorbidades pode ser revertida por meio de tratamento apropriado. Entretanto, aquelas associadas à degeneração musculoesquelética e ao desfiguramento, à hiperplasia de órgãos e a determinados problemas cardiovasculares ainda permanecem como desafios terapêuticos.
REFERÊNCIAS:
MARTIN, Adriana et al. Aplicação do questionário STOP para avaliação de risco de SAHOS em pacientes com acromegalia. Clinical and biomedical research. Porto Alegre, 2015.
FARIA, Carolina et al. Um caso de acromegalia: ver para além do óbvio. Revista Portuguesa de Endocrinologia, Diabetes e Metabolismo, 2016.
SENGER, Maria Helena. Acromegalia: fragmentos históricos e algumas curiosidades. Revista da Faculdade de Ciências Médicas de Sorocaba. ISSN eletrônico 1984-4840, v. 9, n. 3, 2015.
BRASIL. Portaria SAS/MS nº 199, de 25 de fevereiro de 2013, republicada em 22 de novembro de 2013. Protocolo Clínico e Diretrizes Terapêuticas da Acromegalia. Diário Oficial da República Federativa do Brasil, Ministério da Saúde, Brasília, DF no 39, de 27 de fevereiro de 2013, Seção 1, página 113.



